Arkaitz Mucientes1, Martín-Núñez Gracia María2, José Lisbona-Montañez3, Patricia Ruiz-Limon4, Rocío Redondo-Rodríguez5, SARA MANRIQUE6, Inmaculada Ureña5, Laura Cano-García5, Isabel Moreno-Indias1, Natalia Mena Vazquez7 and Antonio Fernandez-Nebro8, 1IBIMA Plataforma BIONAND, Málaga, Spain, 2Universidad de Málaga, Departamento de Medicina y Dermatología, Málaga, Spain, 3Universidad de Málaga, Departamento de Medicina y Dermatología, Moclinejo, Spain, 4IBIMA Plataforma BIONAND, Cordoba, Spain, 5Division of Rheumatology, Hospital Regional Universitario Carlos Haya, Málaga, Spain, 6Division of Rheumatology, Hospital Regional Universitario Carlos Haya, Malaga, Spain, 7IBIMA, Málaga, Spain, 8Hospital Regional Universitario de Málaga, Malaga, Spain
Background/Purpose: The etiology of rheumatoid arthritis (RA) is not fully understood. It is accepted that RA results from the interplay of genetic and environmental factors. Epigenetic modifications could serve as a bridge between genetic and environmental factors, potentially influencing the onset and progression of RA. Purpose:to identify differential DNA methylation patterns across the entire genome in patients with RA compared to healthy controls, and to explore epigenetic alterations that couldbe predictors of increased disease severity.
Methods: In a cross-sectional study of a prospective cohort, we examined 64 subjects, including 16 with severe RA, 16 with non-severe RA, and 32 healthy controls. The severity phenotype was determined based on the average of moderate-to-high inflammatory activity, defined by an accumulated Disease Activity Score(DAS28-ESR) ≥3.2, positivity for rheumatoid factor (RF) and anti-citrullinated peptide antibodies (ACPA), as well as elevated levels of Collinsella aerofaciens (OTU ≥0.15). DNA methylation analysis was performed using the Infinium Methylation EPIC BeadChip (Illumina, San Diego, CA, USA), and the methylation level of each cytosine was expressed as a β-value. Descriptive analysis, ANOVA, and bivariate analysis were conducted for statistical comparisons among groups. Additionally, two multivariate logistic regression models were employed to identify factors associated with RA and the severe RA phenotype.
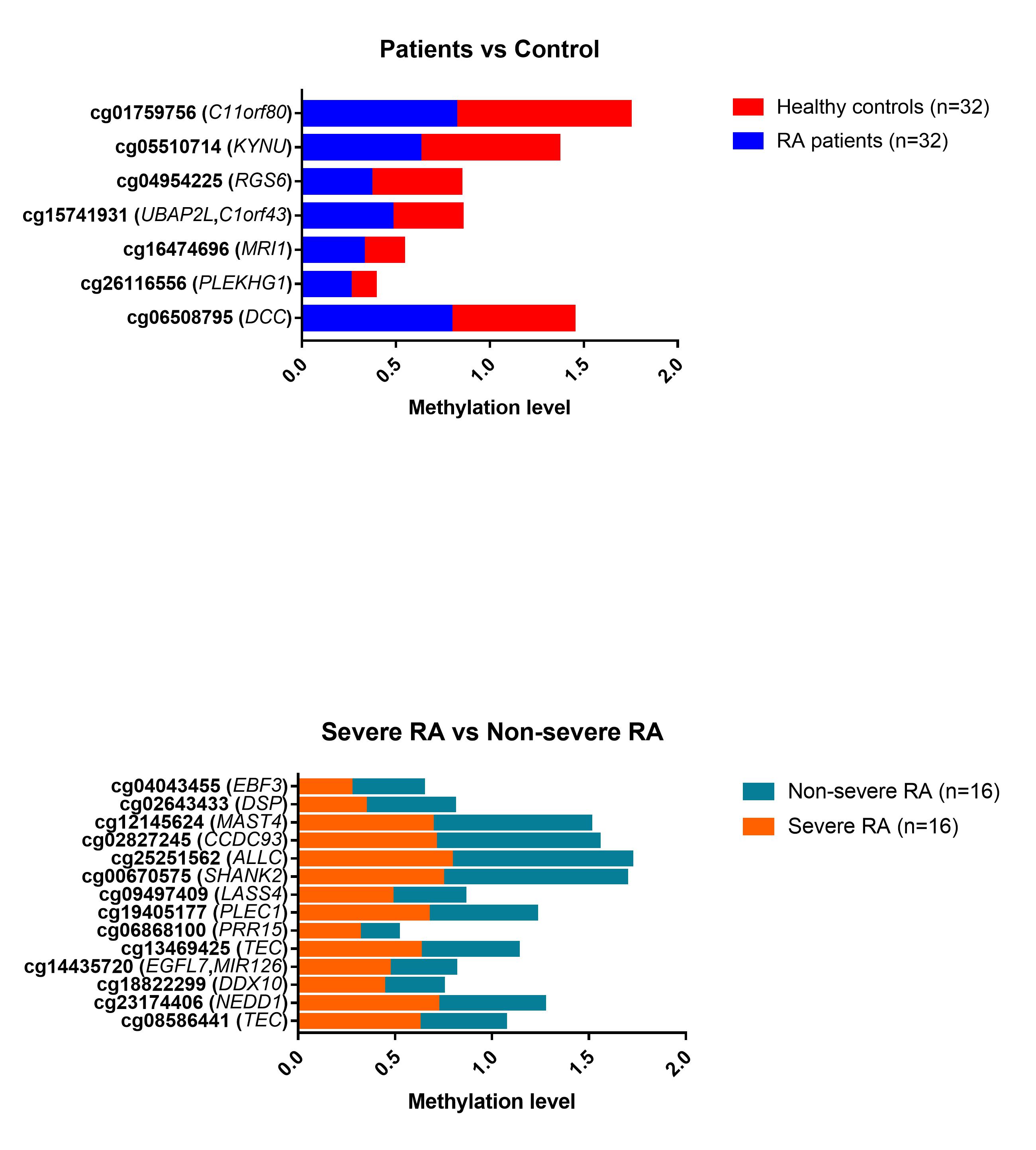
Results: Table 1 shows the main demographic characteristics. The majority (75%) were female, with a mean age of 57.6 ± 9.4 years. RA patients, in comparison with the controls, presented higher smoking habits (62.5% vs 40.7%; p=0.015) and obesity (53.1% vs 28.1%; p=0.012). Among patients, subjects with severe RA compared with non-severe had a higher mean DAS28-ESR (3.9 ± 0.6 vs. 3.3 ± 0.2 mg/L, p=0.001), higher median (IQR) abundance in Collinsella( 0.3 [0.1-1.7] vs. 0.1[0.0-0.4] mg/L, p=0.003), higher frequency of erosions (93.8% vs 50.0%; p=0.006), elevated ACPA (68.8% vs 31.1%; p=0.034) and treatment with biological therapy (56.3% vs 12.5%, p=0.009). Regarding the methylation analysis, among all the differentially methylated CpGs, only those CpGs associated with genes or pseudogenes, exhibiting a minimum β value change of ± 0.10 between groups and a p-value ≤ 0.01, were selected;thus, 7 CpG sites differentially methylated between RA and controls and 14 CpG sites differentially methylated between severe RA and non-severe RA were selected (Figure 1). The CpG sites described in each gene, located in differentially methylated regions, together with other CpGs, were proposed as possible biomarkers (Table 1). In addition, cg06166490 was considered since it is located in a differentially methylated region (adjustedp-value < 0.00001). Of these CpGs, 5 CpG sites were associated with the presence of RA in multivariate model 1 and 2 CpG sites with disease severity in multivariate model 2 (Table 2).
Conclusion: DNA methylation levels at specific CpG sites are associated with RA. The present study identified epigenome marks related to RA and possible disease severity, which warrant further investigation and could be useful in the diagnosis and management of the disease.
Table 1: Demographic and clinical features of the cohort
Table 2: CpG sites selected as possible potential biomarkers
Figure 1: CpG sites with significantly differential methylation between patients with rheumatoid arthritis (RA) and healthy controls (upper panel), as well as between severe and non-severe RA patients (lower panel). The gene symbol is indicated in parentheses for each CpG site.
A. Mucientes: None; M. Gracia María: None; J. Lisbona-Montañez: None; P. Ruiz-Limon: None; R. Redondo-Rodríguez: None; S. MANRIQUE: None; I. Ureña: None; L. Cano-García: None; I. Moreno-Indias: None; N. Mena Vazquez: None; A. Fernandez-Nebro: None.



.jpg)
.jpg)