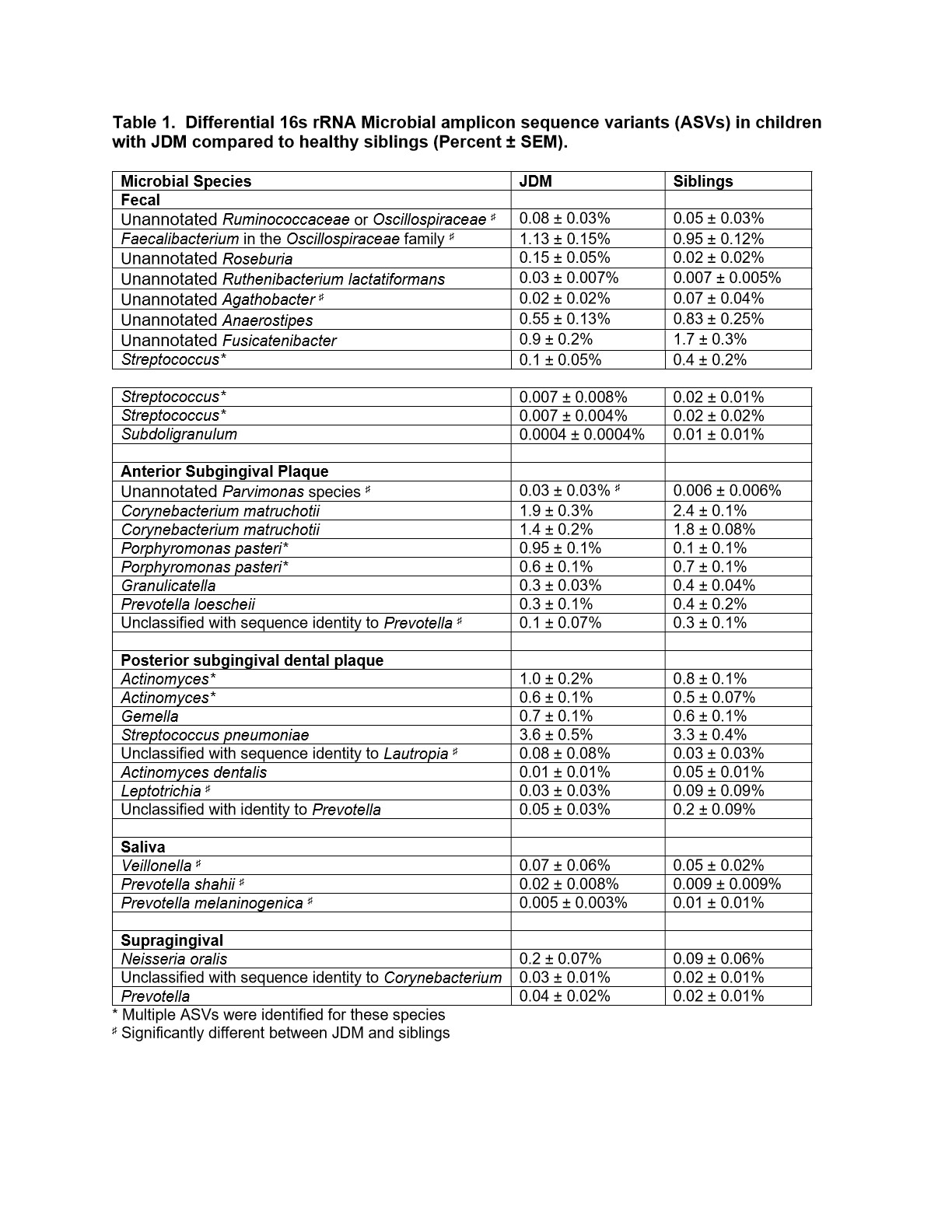
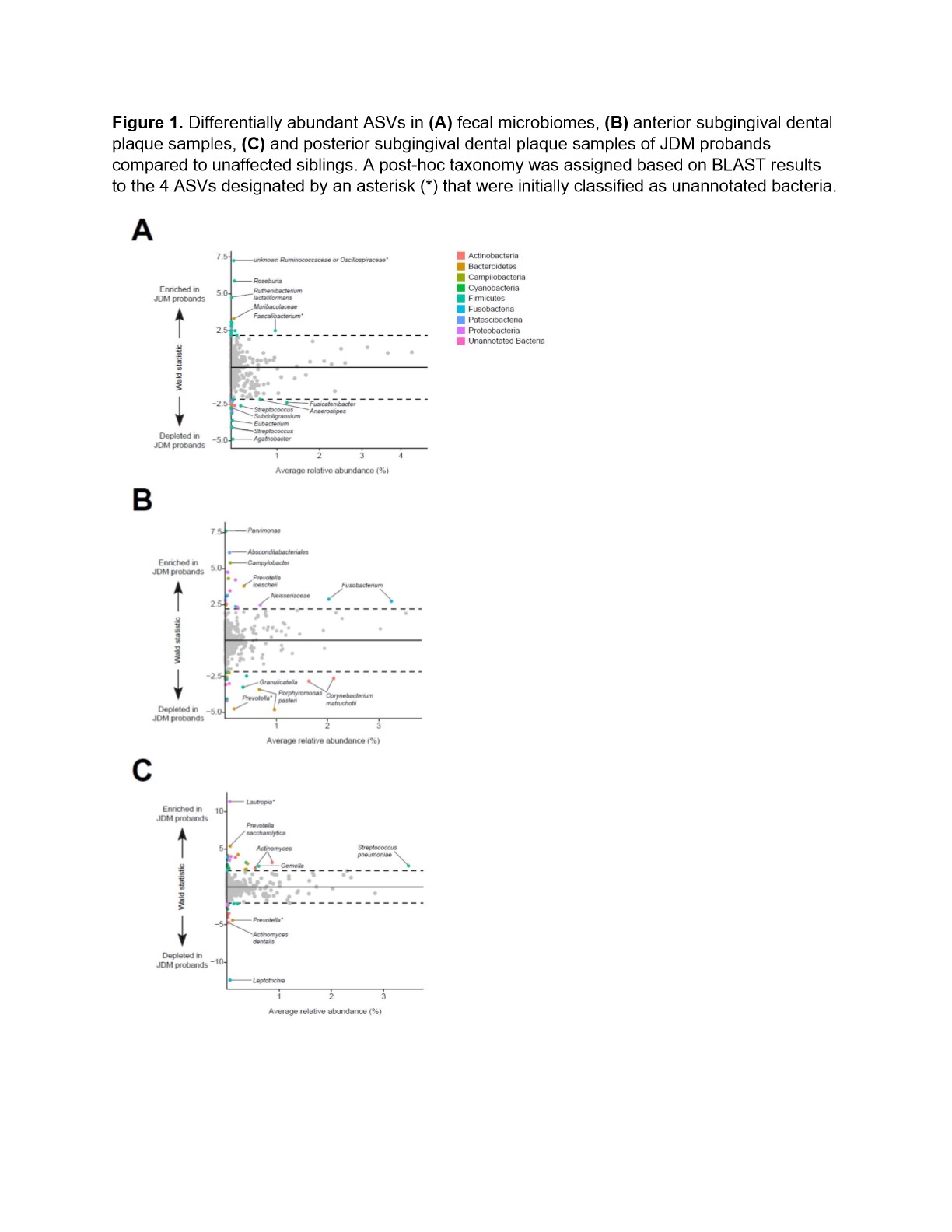
Poster Session B
Pediatric autoimmune diseases: Kawasaki disease, juvenile dermatomyositis and juvenile localized scleroderma


Albert Chow, MD (he/him/his)
Loma Linda University
Loma Linda, CA, United States
Disclosure information not submitted.