University of Michigan Ann Arbor, MI, United States
Disclosure information not submitted.
Bruna Mazetto, NaveenKumar Somanathapura, Claire Hoy, Christine Rysenga, Srilakshmi Yalavarthi, Cyrus Sarosh, Caroline Ranger, Katarina Kmetova, Jacqueline Madison, Yu Zuo and Jason Knight, University of Michigan, Ann Arbor, MI
Background/Purpose: Inappropriately amplified inflammatory responses are hallmarks of many diseases, with extracellular ATP often playing a central role in the orchestration of inflammation. Regulated cellular ATP release is mainly through selective anion channels such as pannexin-1 (PANX1) channels. When cells are under stress, PANX1 channels may become constitutively opened after the cleavage of the C-terminal regions of channel subunits. While hyperactivated platelets contribute to at least a subset of antiphospholipid syndrome (APS)-associated thrombotic events, the most effective way to restrain their activation has remained elusive. Here, we aimed to evaluate the potential role of platelet PANX1 channels in the pathophysiology of APS.
Methods: Extracellular ATP release was evaluated in platelets freshly isolated from patients with positive antiphospholipid antibodies and features of APS (n=51) or from healthy controls (n=16) by standard assay kits and flow cytometry. No patients in this study had concomitant lupus. PANX1 inhibitors included carbenoxolone (CBX) and probenecid (PRB). In some experiments, platelets were stimulated with standard platelet agonists (thrombin, convulxin, and U46619) or with IgG purified from triple-positive APS patients or healthy controls. The participation of purinergic receptors in ATP-mediated platelet activation was assessed with specific inhibitors of various relevant P2X and P2Y purinergic receptors.
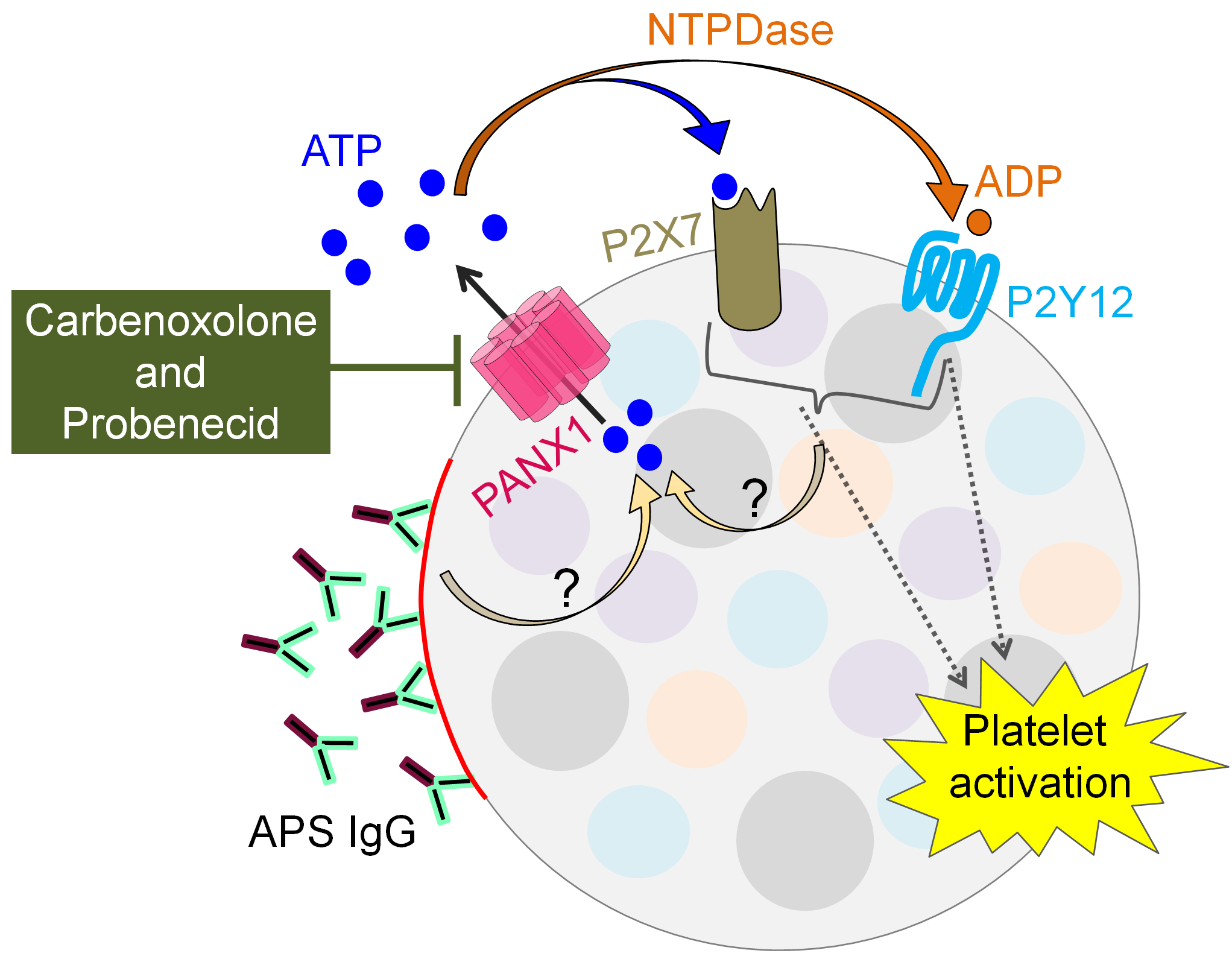
Results: The basal release of ATP from APS platelets was significantly higher than from healthy platelets (median 4-fold, p< 0.0001). This ATP release was strongly reduced in the presence of CBX (2-fold reduction, p< 0.05). Although the difference between APS and healthy platelets was obscured upon activation with thrombin, ATP release in this context was still prevented by either CBX (3-fold reduction, p< 0.0001) or PRB (3-fold reduction, p< 0.0001). Beyond thrombin, the PANX1 inhibitors also significantly reduced platelet ATP release in response to the thromboxane mimetic U46619 (2-fold reduction, p< 0.05). Interestingly, treatment with APS IgG was even more effective than the standard platelet agonists in opening PANX1 channels (4-fold, p< 0.0001) in just 30 minutes. This effect was completely neutralized by CBX treatment (4-fold decrease, p< 0.01). Notably, PANX1 channel-dependent ATP release was blunted by blocking either P2Y12 (p< 0.0001) or P2X7 (p< 0.0001) receptors, suggesting self-perpetuating purinergic pathways for PANX1 activation.
Conclusion: These data highlight the potential role of platelet PANX1 channels in contributing to vascular inflammation in APS by excessive extracellular ATP release. The accumulation of extracellular ATP promotes even further ATP release (and likely platelet activation) via P2Y12 and P2X7 receptors. PANX1 channel blockers such as CBX and PRB could be novel ways to restore platelet homeostasis in APS, potentially restraining thrombosis without a major impact on hemostasis (Figure 1). Experiments are now underway to further evaluate the upstream and downstream signaling partners of activated PANX1 channels in APS platelets.
Illustration of platelet PANX1 channel and downstream purinergic receptors in APS.
B. Mazetto: None; N. Somanathapura: None; C. Hoy: None; C. Rysenga: None; S. Yalavarthi: None; C. Sarosh: None; C. Ranger: None; K. Kmetova: None; J. Madison: None; Y. Zuo: None; J. Knight: Jazz Pharmaceuticals, 2.