AMPEL BioSolutions Charlottesville, VA, United States
Disclosure information not submitted.
Erika Hubbard1, Prathyusha Bachali2, Kathryn Kingsmore Allison1, Amrie Grammer1 and Peter Lipsky1, 1AMPEL BioSolutions, Charlottesville, VA, 2AMPEL BioSolutions, Redmond, WA
Background/Purpose: We previously developed a novel machine learning (ML) pipeline leveraging analysis of gene expression data to identify subsets of SLE patients with common molecular patterns of disease, endotypes[1]. These molecular subsets had significant differences in clinical characteristics, the frequency of subsequent flares, and clinical responsiveness to a lupus biologic, tabalumab. The current study makes use of the ML classifier to determine endotype membership in an independent validation cohort of SLE patients.
Methods: Gene expression by RNA-sequencing of whole blood and clinical metadata were collected from 91 SLE patients from two clinical trials (NCT03626311 and NCT03180021). Patients met ACR classification criteria of SLE and patients from one trial had renal biopsies at the time gene expression was measured. A random forest classifier trained on 2183 lupus patient gene expression profiles was used to predict endotype membership of the 91 patients. Lupus Cell and Immune Score (LuCIS), a continuous score measuring the extent of modular immunologic abnormalities determined by ridge-penalized logistic regression, was also calculated for each patient.
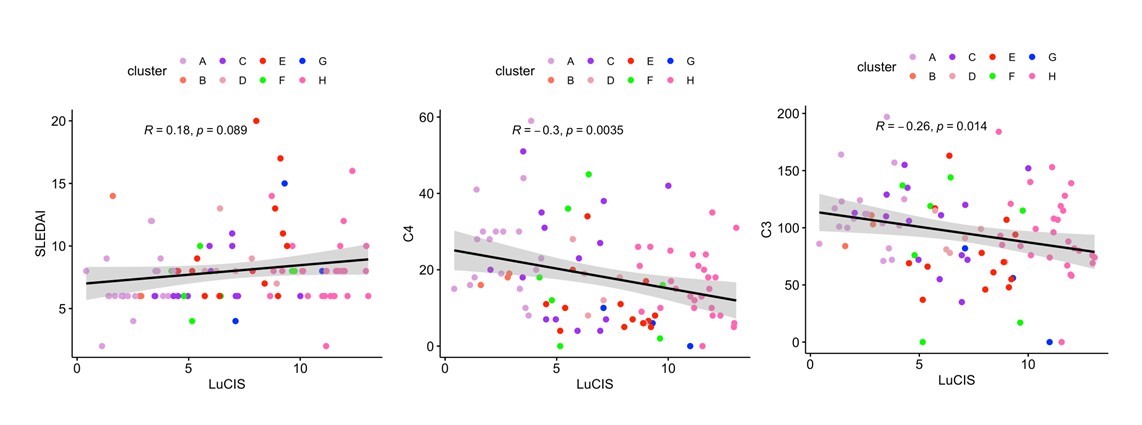
Results: The ML prediction of independent SLE patients into endotypes yielded eight subsets with molecular patterns mirroring those found previously in a development and testing cohort of 3166 patients (Figure 1). Endotypes were designated A-H, with A representing the group with the least number of transcriptional lupus-related aberrancies and H representing the group with the greatest immunologic perturbations. Groups H, A, C, and E were comprised of the greatest number of patients whereas B and G were small and underrepresented in this cohort. Endotype H contained the greatest number of patients with proliferative lupus nephritis (LN) whereas no patient with LN was found in subset A or B. Serum complement differed among the subsets, with the more immunologically active having lower levels. LuCIS values reflected the immunological activity of the subsets but did not correlate with SLEDAI, although they were moderately, inversely correlated with serum C3 and C4 levels (Figure 2). Eight patients had moderate/severe flares during the six months of the trials, of whom all had elevated LuCIS scores at baseline.
Conclusion: A novel endotyping pipeline based on transcriptional profiles and ML accurately identified patient endotypes in new datasets. Patients in the endotypes with the least immunologic activity did not have proliferative nephritis and also experienced no lupus flares during the subsequent six months. Endotyping SLE patients based on gene expression profiles can provide important prognostic information and provide novel molecular insights in support of personalized management.
1. Kim YH, Park MR, Kim SY, Kim MY, Kim KW, Sohn MH. Respiratory microbiome profiles are associated with distinct inflammatory phenotype and lung function in children with asthma. J Investig Allergol Clin Immunol. 2023 Jun 1:0. doi: 10.18176/jiaci.0918. Epub ahead of print. PMID: 37260034.
Figure 1. Identification of Endotypes Among 91 SLE Patients Molecular subsets identified by a random forest algorithm using gene set variation analysis (GSVA) enrichment scores of 26 immune/inflammatory modules. Clinical metadata for each patient (x-axis) was annotated as shown. Heatmap constructed in R using the ComplexHeatmap package.
Figure 2. LuCIS Correlations with Clinical Data Pearson correlations of LuCIS values with baseline clinical characteristics. Each data point is colored by endotype membership. Plots were constructed in R using the ggplot2 package.
E. Hubbard: None; P. Bachali: None; K. Kingsmore Allison: None; A. Grammer: None; P. Lipsky: None.



.jpg)