University of Mysore, Karnataka, India Ann arbor, MI, United States
Disclosure information not submitted.
NaveenKumar K. Somanathapura, Claire Hoy, Srilakshmi Yalavarthi, Cyrus Sarosh, Bruna De Moraes Mazetto Fonseca, Caroline Ranger, Christine Rysenga, Ajay Tambralli, Jacqueline Madison, Yu Zuo and Jason Knight, University of Michigan, Ann Arbor, MI
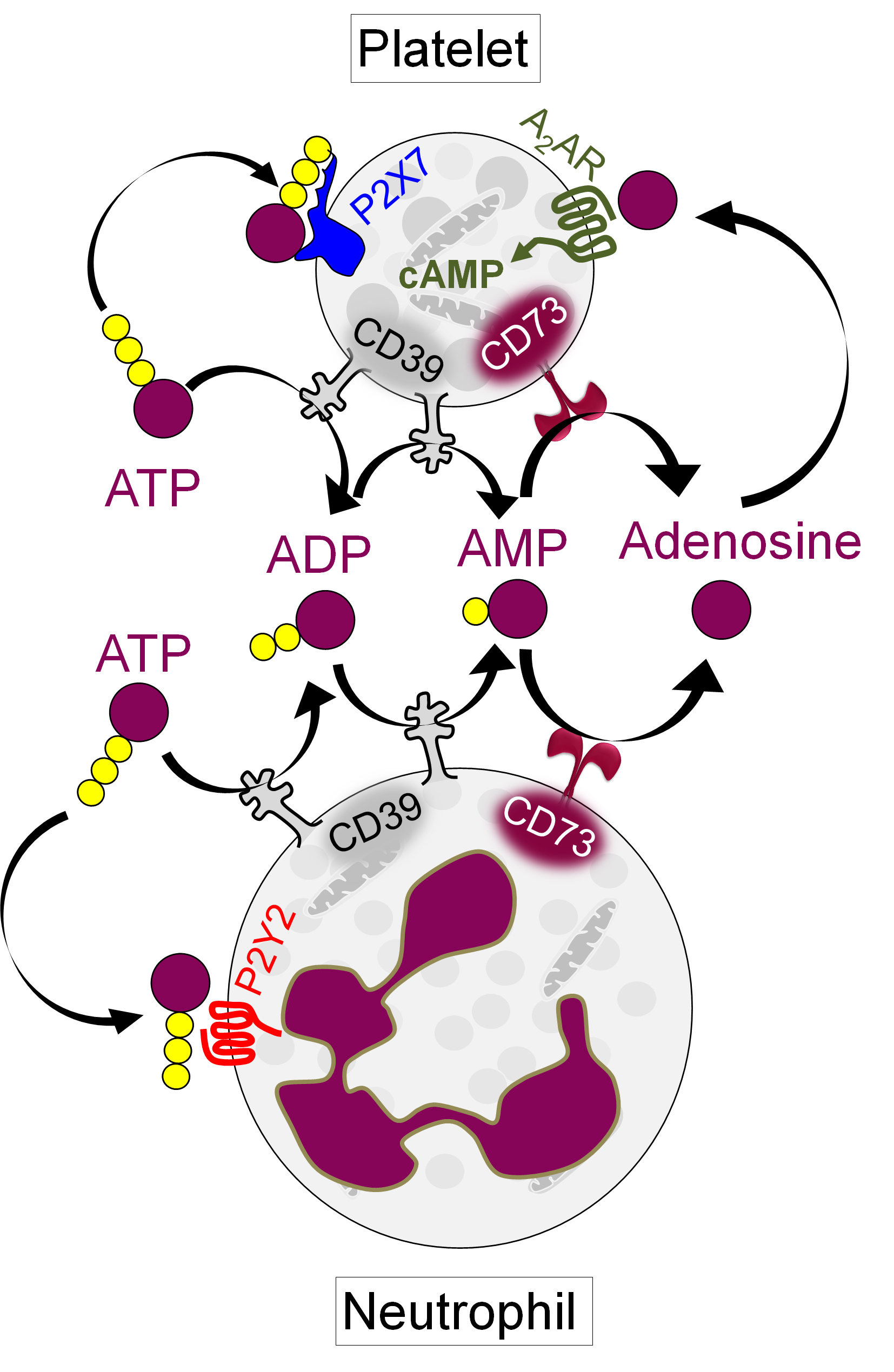
Background/Purpose: In first responding cells such as neutrophils and platelets, extracellular ATP released from activated or dying cells engages cell-surface receptors to launch proinflammatory and prothrombotic signals. As a counterpoint to this thromboinflammatory purinergic signaling, the surface ectonucleotidases CD39 (ATP to AMP) and CD73 (AMP to adenosine) convert ATP into homeostatic adenosine. Adenosine then activates G protein-coupled receptors to increase intracellular cyclic AMP (cAMP), thereby blunting inflammation and thrombosis. Here, we aimed to understand the potential relationship between the CD39/CD73 axis, neutrophils, and platelets in antiphospholipid syndrome (APS).
Methods: Neutrophil-platelet aggregates (NPAs) and platelet P-selectin (CD62P) were assessed in the blood of patients with primary APS by flow cytometry. In parallel, CD39 and CD73 activities on both neutrophils and platelets were measured by a standard malachite green assay. Levels of adenosine generation and intracellular cAMP were estimated using standard kits. In some experiments, healthy neutrophils and platelets were treated with APS IgG, specific CD39/CD73 activity inhibitors (ARL 67156, PSB 12379), or inhibitors of various surface receptors.
Results: As compared with healthy controls (n=48), patients with primary APS (n=55) showed at least a 50% reduction in median activities of neutrophil CD39 (p< 0.0001), neutrophil CD73 (p< 0.0001), platelet CD39 (p< 0.0001), and platelet CD73 (p< 0.0004). These changes were negatively correlated with significant increases in both NPAs (up to 80%, p< 0.0001) and platelet activation as defined by CD62P expression (p< 0.002). The levels of NPAs were higher in patients with APS who had a history of thrombosis than those without. When healthy neutrophils and platelets were cultured with either APS IgG or a CD39 inhibitor, there was a dose-dependent increase in NPA formation (from baseline 15% to 70%), and this phenotype was substantially blocked by inhibition of either the neutrophil P2Y2 receptor or the platelet P2X7 receptor. Focusing further on APS platelets, we found a significant decrease in their ability to generate adenosine (p< 0.01), as well as in their accumulation of intracellular cAMP (p< 0.001). Notably, CD62P expression was inversely correlated with platelet cAMP levels in patients (r=-0.52, p=0.007). Exposure of healthy platelets to APS IgG induced AKT phosphorylation (ser473) followed by downstream activation of GSK3β (ser9). This pro-activation signaling was efficiently blunted by agents that either activated adenosine receptors or directly boosted intracellular cAMP.
Conclusion: In primary APS, deficiency of CD39 and CD73 on both neutrophils and platelets potentiates pro-thrombotic platelet activation and NPA formation. By interrogating the downstream mechanisms, we identified several potential therapeutic targets including neutrophil P2Y2 and platelet P2X7 (Figure 1). Adenosine receptor agonists might be a strategy for restoring platelet homeostasis in APS. Overall, we speculate that a subset of patients with APS would benefit from therapies that modulate extracellular purinergic signaling.
Figure 1: Schematic representation of purinergic signaling in platelets and neutrophils.
N. Somanathapura: None; C. Hoy: None; S. Yalavarthi: None; C. Sarosh: None; B. De Moraes Mazetto Fonseca: None; C. Ranger: None; C. Rysenga: None; A. Tambralli: None; J. Madison: None; Y. Zuo: None; J. Knight: Jazz Pharmaceuticals, 2.